The human world is in the throes of a deadly coronavirus outbreak, yet it’s just one among tens of thousands of coronaviruses in the animal kingdom.
Very few of them cross over to humans. So how do we dig through the mountain of coronaviruses and find which ones could potentially spill over? Where could the next pandemic come from?
It’s a puzzle that Michael Letko, virologist and assistant professor at Washington State University, has pondered for years. Now, he and his colleagues have found a way to identify coronaviruses likely to affect people.

(Courtesy Nature Research Microbiology Community)
A focus on coronaviruses makes sense. At least four separate and severe outbreaks that affected humans and livestock emerged from coronavirus spillover events: SARS-CoV in 2003, MERS-CoV in 2012, SADS-CoV in 2018 (a devastating porcine virus), and now SARS-CoV-2 (the cause of COVID-19).
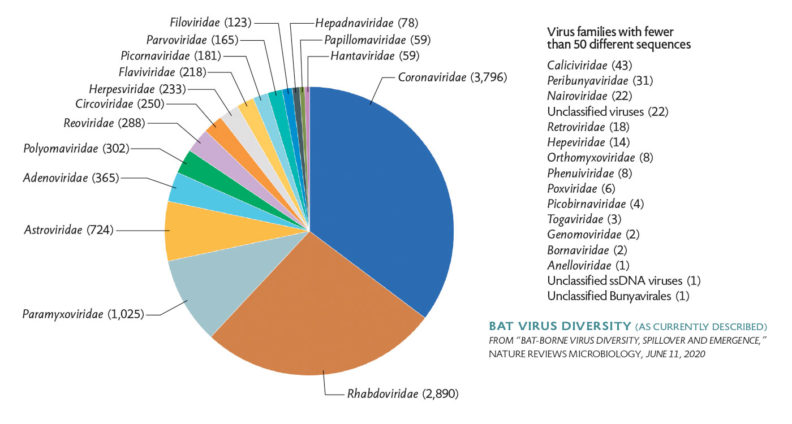
The prevalence of coronaviruses also makes it a promising candidate for zoonotic outbreaks because they thrive throughout animal species, especially in bats.
“While many coronaviruses have been discovered in bats, coronavirus sequences have now been reported in hedgehogs, whales, chickens, pigs, and seals; pick an animal and it probably has a coronavirus,” Letko wrote for Nature’s microbiology blog.
Bats, though, have been identified as hosts for several emerging diseases dangerous to humans. They’re the second most diverse mammalian order, found on every continent except Antarctica, and have a high viral diversity relative to other mammals.
While there is a lot we don’t know about bat ecology and their ability to resist viruses, we do know that several bat viruses have a high similarity to SARS-CoV-2.
At the molecular level, says Letko, lineage B betacoronaviruses, such as SARS, have receptor areas on spike proteins that interact with people’s cells. These areas are nearly identical between various bat species, intermediate species, and humans.
Armed with that information, Letko began to narrow down the search for potentially dangerous coronaviruses.
The advent of advanced genetic sequencing in the early 2000s led to a boom in identifying the genomes of coronaviruses. There are over 40,000 results for the search term “coronavirus” on the public Genbank database.
The problem, says Letko, is not enough downstream analysis of that information. “We actually have not studied 99.9% of these viruses in the laboratory,” he wrote. “Therefore, we do not know if these viruses that we are constantly discovering can cross the species barrier to humans, if they can transmit to our livestock, or if they can cause disease.”
Using well-known techniques of synthesizing and analyzing DNA, Letko and his team concentrated on just a small portion of the coronavirus: the receptor binding domain.
“It’s focusing on the smallest and most important things we can,” says Letko.
Since they only test a small region of the virus, it’s faster and costs much less than examining the entire virus, which means they can scale up and test large numbers of viruses.
Before coming to WSU, Letko worked at the National Institutes of Health laboratory in Hamilton, Montana, where Letko and his team at the NIH put their system to the test in early 2020. When reports came from China about a new coronavirus around Wuhan, the researchers were able to quickly demonstrate the host receptor of SARS-CoV-2 within just 12 days.
Letko’s new lab at WSU’s Paul G. Allen School for Global Animal Health aims to be part of a global network of labs, EcoHealth, focused on functional viromics.
“The ultimate goal of this work is to move away from the rush to learn basic virus biology after virus spillover and emergence…” wrote Letko.
As Letko and others build on the knowledge of viruses, the world could move from reaction to prediction of those pathogens with the greatest threat.
 Click on above chart for PDF enlargement
Click on above chart for PDF enlargement