
Guy Palmer (’84 Ph.D.) and Terry McElwain (’86 Ph.D.) are on the hunt. The Washington State University research veterinarians know their quarry—Anaplasma and Babesia, the pathogens that cause two of the world’s most debilitating diseases of livestock—but they haven’t found the right weapon to bring them down. What they need, what they have worked for years to find, are vaccines that will stop the pathogens dead in their tracks.
Vaccines are such a routine part of health care for us that they can seem like old news. Because of effective vaccines, those of us who live in developed countries don’t have to worry about measles, polio, or smallpox, diseases that inflicted grave harm on our grandparents’ generation. Other than “new” diseases like next year’s strain of influenza, we’re well protected. Yet, some of the most common infectious diseases of people and animals have been around just about forever—and we still don’t have vaccines against them.
“The way I look at this is that we have vaccines for all the ones that are easy to vaccinate against,” says McElwain. They’re easy because of their biology, he says. When you get the disease, it either kills you or your immune system fights it off and the pathogen is cleared from your system. Vaccination gives you a head start in the fight; it primes your immune system to recognize and get rid of the pathogen before harm is done.
The “hard” diseases operate differently. If you get infected with one of these pathogens, you will probably remain infected for life. Your immune system can’t get rid of it. In humans the list of such diseases includes malaria, sleeping sickness, and syphilis. In cattle, it includes anaplasmosis and babesiosis, the diseases Palmer and McElwain have targeted. Both are tick-borne blood infections that cause severe anemia, often leading to death, and both share an interesting MO.


“It’s an impressive disease to see,” says McElwain of babesiosis. “Animals go from being fairly normal-looking one day to just very, very sick the next.” Anaplasmosis is usually less dramatic, but it is more widespread, affecting more than two-thirds of the cattle in some regions. The economic costs of the diseases are enormous—billions of dollars a year in lost animals and lowered productivity—but the human costs are immeasurable. In sub-Saharan Africa, for instance, smallholder farmers depend on their small herds of cattle or goats for food, for cash income, for status, and as beasts of burden.
“The loss of one animal can have a profound effect on the family’s well-being,” says McElwain. “It may be the difference between one of the children going to school or not.”
Palmer says most persistent pathogens are transmitted by vectors (ticks, mosquitoes, tsetse flies) or by sexual contact. Since they can’t spread to new hosts by casual contact, the pathogens have to survive in one host until a transmission opportunity comes along. Unfortunately for us, the strategies they’ve evolved to avoid the host’s defenses also stymie our efforts to make a vaccine.
A vaccine works by showing the body’s immune system a pathogen or part of a pathogen (usually a protein, in this context called an antigen) so that it can develop cellular memory and antibodies that will recognize and attack the pathogen in the future.
Many vaccines use the entire pathogen, which has been killed or weakened so it won’t cause the full-blown disease. Such vaccines work against persistent pathogens, but they are often expensive to make and difficult to deploy. Live vaccines, for instance, need a “cold chain”: they must be kept cold or frozen until just before use, which is a tall order in poor countries in the tropics. A subunit vaccine, based on just one or a few proteins, is usually cheaper and hardier. Theoretically, any protein that sparks an immune response and is a distinctive feature of the pathogen could be the basis for a good vaccine. The problem is that no one’s been able to make a subunit vaccine against Anaplasma, Babesia, or any of the other persistent pathogens.
“Our challenge is to figure out why that is,” says McElwain. “Biologically and immunologically, why is that the case? If we could get a handle on that, I think we’d really have a significant advance.”
In the mid-1980s, a flurry of new techniques for working with proteins and DNA led to identification of a key malaria antigen. Palmer and McElwain, just beginning their careers on the WSU faculty, immediately applied the new techniques to their organisms.
When Palmer used the new tools to search for Anaplasma antigens, he found Msp2 (major surface protein 2). Msp2 is the most abundant protein on the surface of the pathogen. Both ends of Msp2 are embedded in the Anaplasma cell membrane, and the middle portion loops out into the extracellular space. Since the loop is exposed to the bloodstream, whenever Anaplasma goes looking for new red blood cells to infect, it is the part of the protein the host’s antibodies have a chance to recognize and grab onto.
In other words, Msp2 looked like a perfect candidate as a vaccine antigen.
Then why doesn’t the host’s immune system target Msp2 and knock out the infection? That part still didn’t make sense. Palmer’s lab found that infected cows do make antibody against Msp2, a lot of it; that’s likely what drives down the number of pathogen cells and allows the cow to recover from acute illness. But why do pathogen numbers rise again a couple of weeks later? Why doesn’t the cow clear the infection completely?
After months of protein and gene analysis, Palmer’s team reached a stunning conclusion. The pathogen persists because the host no longer recognizes it—because Msp2 changes. It’s still present, but it doesn’t look like the Msp2 that was there before. Palmer’s group found that at any given stage of the infection, several forms of Msp2 are present—and none of them are recognized by antibodies the host made in earlier stages. By altering the most abundant protein in its surface coat, Anaplasma avoids detection by antibodies the host has already made.
“That’s why the immune system can never catch up with what’s going on,” says molecular biologist Kelly Brayton. She describes Msp2 as throwing up a smokescreen that allows Anaplasma cells to escape direct attack by the immune system. “Part of the job of this molecule is to be this thing that’s going, ‘Hey! Look at me!’ Because it can change. So it’s trying to attract the attention of the immune system, and then as soon as the host makes a response to that particular variant, it’s moved on.”
So how did Anaplasma do it?
Palmer calculated that a cow infected early in life might see more than 1,000 variants of Msp2 over its lifetime. The Anaplasma genome codes for fewer than 1,000 proteins. There’s no way it could have 1,000 genes for Msp2 alone. The pathogen had to be doing something unusual with its genes to generate that many variants. Brayton sequenced the entire Anaplasma genome and looked for something that would explain the diversity of Msp2 forms. What she found is a process called “gene conversion,” a masterwork of deception in which a handful of “pseudogenes” mix and mingle to continually change the identity of Msp2—and of Anaplasma itself.
It was a major finding. Gene conversion explained how Anaplasma escapes detection by the immune system. It also led to the discovery, by researchers elsewhere, that a similar process occurs in the pathogen that causes relapsing fever in humans.
And yet, figuring out how gene conversion works was a bit like finally bursting into the room of a con man you’ve been hunting for years, to find only a trunk full of wigs and false noses. The team still wasn’t in sight of a vaccine. And the culprit himself had fled again.
So Anaplasma disguises itself. Can we help the immune system see past the Msp2 mask? Is there some other surface protein—one that doesn’t change every few weeks—that could be used as a vaccine? Msp2 garners most of the attention from the immune system, but there are dozens more, present in small amounts and not well understood. Most intriguing of all, vaccinating with Msp2 alone does not protect against Anaplasma infection—but vaccinating with a mixture of outer membrane proteins does.
“So there’s something in the outer membrane that’s important,” says immunologist Wendy Brown. Unfortunately, she says, a vaccine made of membrane preparations is “impractical. You’d have to pay $500 a shot, probably.” She set out to discover which proteins in the outer membrane mixture confer protection. Perhaps they could be the basis for an economical vaccine.
Analyzing membrane proteins is not a new idea, but until recently the experiments weren’t feasible. Getting enough of any one protein to use as a test vaccine was difficult, and testing any vaccine in cattle takes months. Testing multiple combinations of proteins was simply unmanageable.
Brown invented a way to test proteins on cow cells rather than in a whole cow. She first immunizes a cow with the mixture of membrane proteins. After the cow’s immune system has had a chance to respond, she takes a blood sample, and from that she extracts T cells. Those are the immune system’s “memory cells”; if the vaccinated cow were to be bitten by an Anaplasma-carrying tick, its T cells would recognize antigen(s) on the Anaplasma surface and would trigger antibody production by other immune system cells. Brown presents the T cells with individual Anaplasma proteins in a way that mimics what happens in the body. If the T cells recognize a protein, they start dividing and making interferon—which tells her the cow’s immune system had responded to that particular protein. It’s a nifty way to rapidly screen a large number of proteins for their potential as vaccines.
With help from WSU chemist Bill Siems, Brown identified more than 20 proteins from Anaplasma‘s outer membrane that had never been described before and that were recognized by T cells from vaccinated cows. Now the group is testing whether any of the proteins make an effective vaccine. They’ve found that a combination of some of the proteins does protect against infection by Anaplasma. Will the mixture lead to a commercially viable vaccine? Brown and her colleagues don’t know yet; but after years of chasing the shape-shifting Msp2, it’s an encouraging result.
Like Anaplasma, Babesia is studded with proteins, at least two of which change over time. Because Babesia‘s genome is larger and more complex than Anaplasma‘s, the team hasn’t yet figured out how the proteins change. The one thing that’s clear is that Babesia doesn’t use the same process of gene conversion as Anaplasma.
In addition to working on variable proteins, Brayton and McElwain have teamed with The Institute for Genomic Research to take a closer look at Babesia‘s genome. They’ve discovered a group of DNA sequences they call “SmORFs,” for “small open reading frames.” (“We took some heat on that name from reviewers, but we stuck to it,” says McElwain. “We’ve got to have some fun with this.”) Since all 44 SmORFs lie next to genes for a variable surface protein, McElwain suspects they’re involved with immune escape, but work on them is still preliminary.
Although its variable proteins remain puzzling, Babesia might be outmaneuvered another way. In the 1960s, scientists in Australia discovered that if they infected a calf with Babesia, let the infection develop for about a week, drew some blood from that calf and used it to infect another calf, and did that 25 to 30 times, the pathogen would lose its ability to cause disease. It still provoked an immune response when inoculated into a new host, but the host no longer got sick; instead, it gained protection. In other words, the so-called attenuated strain worked as a vaccine.
It worked so well that attenuated strains have been used in Australia and Israel—both fertile grounds for Babesia—for many years. Attenuated Babesia vaccines have been banned in many other countries, including the United States, because they are blood-based and might carry other pathogens. For countries like the U.S. where Babesia is not a big threat, they’re not worth the risk. Even in countries where Babesia is a threat, their value is limited because they require a cold chain. They’re also not a permanent solution; every few years, they change so they no longer protect the host. When that happens, the whole attenuation process has to be repeated.
In 2005, the WSU team won a $1.8 million grant from the Wellcome Trust to figure out why attenuated Babesia protects against further infection, yet does not cause disease itself. Can we design strains of Babesia that will mimic the attenuation effect, and that can be used as vaccines? Such a vaccine would avoid the threat of blood-borne pathogens and the tedious and expensive attenuation process.
Colleagues in Argentina have already produced an attenuated strain the old-fashioned way. While they test it on herds there, Brayton and molecular biologist Audrey Lau in Pullman are comparing the genome of the attenuated strain with that of the original strain. Since they know the sequence of the original, they can trace any changes that occurred during the attenuation process. With luck, they’ll identify what change(s) turned a killer into a life-preserving vaccine.
“The clearest-cut scenario is that a gene’s missing,” says Brayton. “You’re usually not that lucky. But [if that happens], you could [ask], what is that gene in the virulent strain? What do we know about it?” Even if nothing is missing, they might find that one or more genes have changed. If Lau and Brayton find clear differences between the strains, the next step will be to create a copy of the attenuated pathogen in the lab and test its effectiveness as a vaccine.
The field tests will be done by the group’s collaborators in Mexico and Argentina, working with herds under natural conditions. Palmer says translating lab results into real-world applications is central to what the WSU group is trying to do—and essential to developing a vaccine that will work against the form of the pathogen that cattle actually encounter.
“People don’t like to do it, because nature’s messy, you know, and the lab is not,” he says. “For a long time there’s been a tendency to rely on strains [of pathogens] that are well established in the laboratory. But when you get out into the field, you find that isn’t necessarily representative of what’s out there in the natural situation.”
No one in the group is predicting when they’ll have a vaccine for either disease. Anaplasma and Babesia have confounded expectations before. But the team members share a sense of excitement about their progress—and an admiration for their elusive adversaries. The most rewarding aspect of their work so far, says McElwain, has been gaining an understanding of the complex, elegant strategies that enable these pathogens to persist in their supposedly more sophisticated hosts.
“That’s been something that we started out, 25 years ago, not appreciating,” he says. “And that is not only something that you can appreciate, if you can find beauty in these organisms. It’s also the challenge, the huge challenge that we have.”
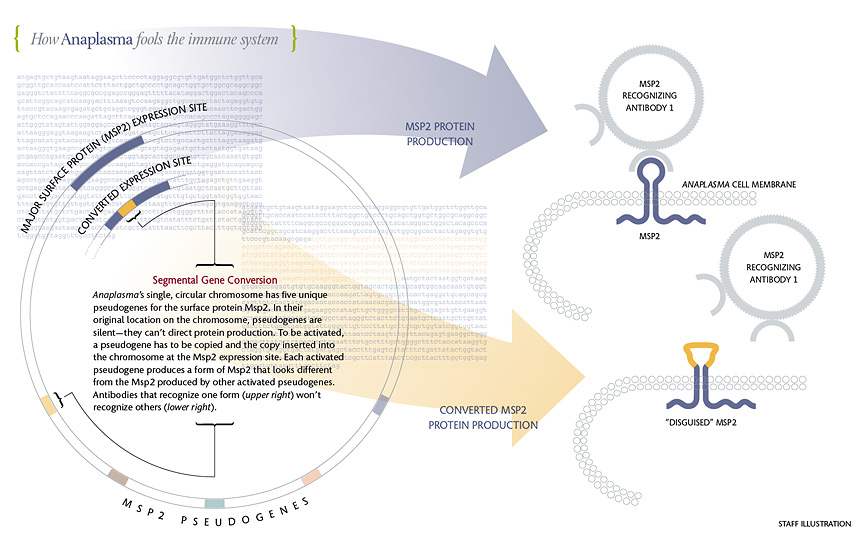 Msp2 changes form when a silent pseudogene is copied and the copy is plugged into the expression site, displacing the pseudogene copy that had been there. Once it is installed in the expression site, a pseudogene is able to create its version of Msp2, whose middle loop differs from the loop in the previous Msp2. Antibodies that recognized the original Msp2 can’t recognize the new form, so Anaplasma cells that are making the new form thrive. It takes the immune system a few weeks to make antibodies against the new Msp2. By that time, another gene conversion event has occurred and a third form of Msp2 is on the scene. If gene conversion involved only whole pseudogenes, Anaplasma would be able to make just five versions of Msp2—not enough to escape attack by the host’s immune system for years. The incredible variety of Msp2 comes about because the pseudogenes can mingle. Guy Palmer and Kelly Brayton found that slivers of up to four pseudogenes can be combined at the expression site. Each combination creates a form of Msp2 that’s different from previous forms—different enough so that antibodies that recognized earlier forms won’t recognize the new one. Palmer estimates that the system enables Anaplasma to make at least 1,024 different versions of Msp2. Click on infographic for larger size
Msp2 changes form when a silent pseudogene is copied and the copy is plugged into the expression site, displacing the pseudogene copy that had been there. Once it is installed in the expression site, a pseudogene is able to create its version of Msp2, whose middle loop differs from the loop in the previous Msp2. Antibodies that recognized the original Msp2 can’t recognize the new form, so Anaplasma cells that are making the new form thrive. It takes the immune system a few weeks to make antibodies against the new Msp2. By that time, another gene conversion event has occurred and a third form of Msp2 is on the scene. If gene conversion involved only whole pseudogenes, Anaplasma would be able to make just five versions of Msp2—not enough to escape attack by the host’s immune system for years. The incredible variety of Msp2 comes about because the pseudogenes can mingle. Guy Palmer and Kelly Brayton found that slivers of up to four pseudogenes can be combined at the expression site. Each combination creates a form of Msp2 that’s different from previous forms—different enough so that antibodies that recognized earlier forms won’t recognize the new one. Palmer estimates that the system enables Anaplasma to make at least 1,024 different versions of Msp2. Click on infographic for larger size